
A fibrose cística, também conhecida como mucoviscidose, ou somente FC, é uma doença genética, hereditária, autossômica e recessiva, ou seja, passa de pai para filho. Se manifesta em ambos os sexos, é grave, sem cura e afeta as glândulas exócrinas, provocando alterações nos pulmões, pâncreas, fígado, intestino e as glândulas sudoríparas. Sua principal característica é o acúmulo de secreções mais densas nessas áreas do corpo.
O gene da Fribrose Cística é transmitido pelo pai e pela mãe (embora nenhum dos dois manifeste a doença) e é responsável pela alteração no transporte de íons através das membranas das células. Isso compromete o funcionamento das glândulas exócrinas que produzem substâncias como o muco, suor ou enzimas pancreáticas. Ou seja, a fibrose cística é causada por um gene que faz com que o corpo produza um líquido anormalmente denso, conhecido como muco, que se acumula nas passagens respiratórias dos pulmões e também no pâncreas.
Essa obstrução por secreção mais espessa também pode acometer os dutos biliares. A bile retida no fígado favorece a instalação de um processo inflamatório.
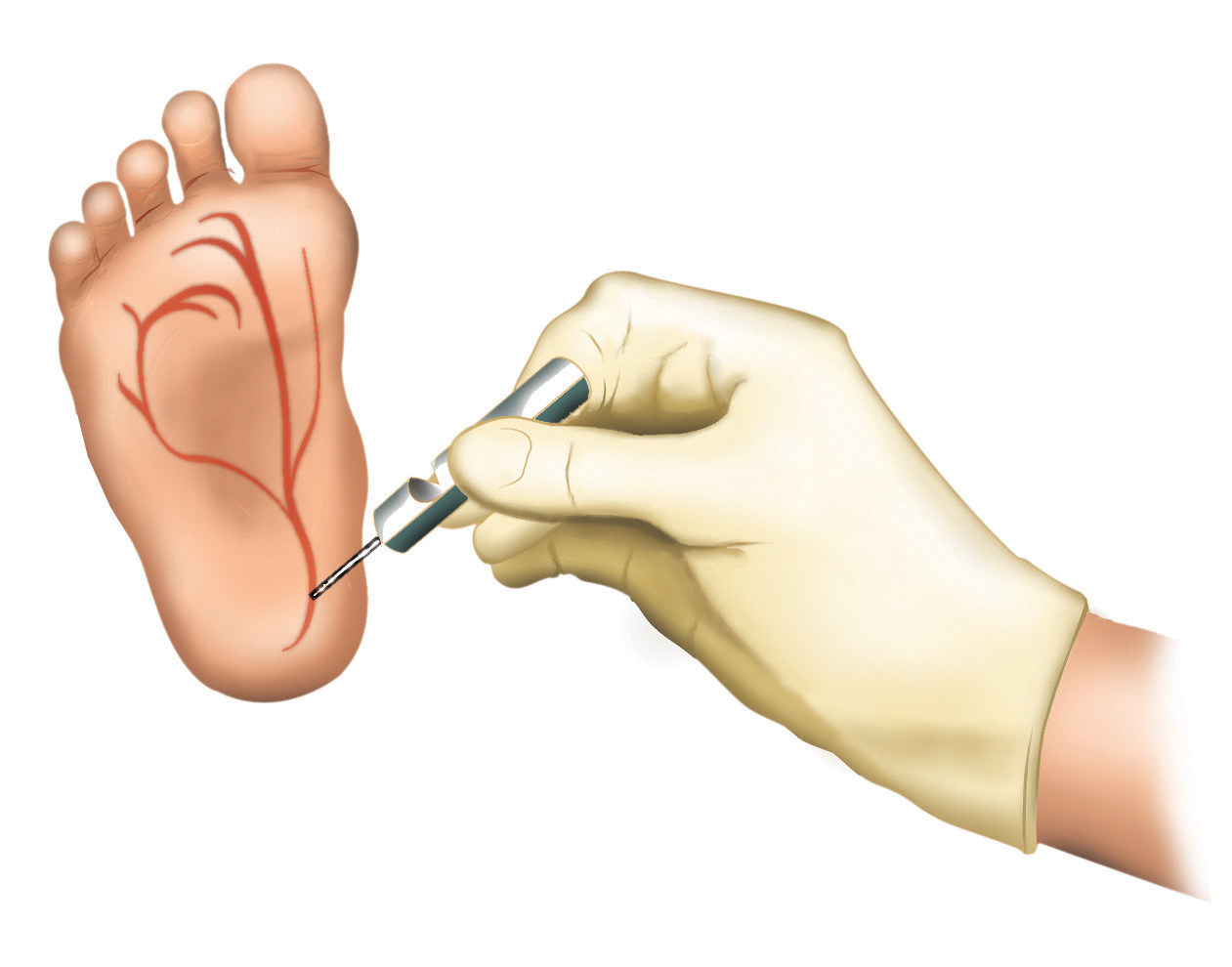
A doença pode ser precocemente identificada através do teste do pezinho e seu diagnóstico pode ser confirmado através do teste do suor ou ainda através de exames genéticos.

Estudos científicos mostram que uma criança com pais portadores assintomáticos, ou seja, que não apresentam sintomas tem 25% de chances de ter a doença, 50% de ser um portador e 25% de não ter a doença.
A maioria das crianças com fibrose cística é diagnosticada até os dois anos de idade. Um número menor, no entanto, só é diagnosticado com 18 anos ou mais.
Ela é mais comum entre caucasianos e é rara nas raças negra e asiática, o que torna um desafio para a comunidade científica mundial o estudo da genética das populações, no sentido de encontrar a cura definitiva.
Uma em cada 25 pessoas (4%) dos descendentes de europeus carreia o gene para fibrose cística. Aproximadamente 30.000 americanos possuem, fazendo desta uma das doenças hereditárias mais comuns.
Na Europa atinge um em cada 5.000 pessoas, nos Estados Unidos e Canadá um em cada 4.000, enquanto no Brasil estima-se que atinge cerca de 1 a cada 10.000, sendo maior incidência no Sul e Sudoeste do país.
As principais complicações dos pacientes são: sangramento pulmonar, infecções graves, pneumotórax (vazamento de ar do pulmão) e obstrução intestinal:
Dificuldade para respirar (92%);
Infecções das vias respiratórias frequentes e persistentes (90%);
Infertilidade (90%);
Inúmeros problemas digestivos (55%);
Menor crescimento e desnutrição (56%);
Obstrução do íleo terminal por um mecônio espesso (6-15%);
O amontoado de muco pode resultar em infecções pulmonares que podem colocar a vida do paciente em risco e podem levar a problemas digestivos graves.A doença pode afetar as glândulas sudoríparas e o sistema reprodutivo masculino.
A obstrução dos dutos pancreáticos pela secreção mais viscosa pode impedir que as enzimas digestivas sejam lançadas no intestino. O paciente assim tem má absorção de nutrientes e não ganha peso, apesar de alimentar-se bem.
Ainda no período neonatal (em recém-nascidos) é possível observar o entupimento do intestino, dificuldade de ganhar peso, tosse com secreção e desidratação sem motivo aparente.
Com o passar dos anos, o paciente pode apresentar perda de peso, desnutrição progressiva, tosse crônica com muita secreção, sinusite crônica, formação de pólipos nasais (aumento da mucosa do revestimento do nariz que forma pseudotumores no nariz), doença hepática (cirrose biliar), diabetes, infecções respiratórias e infertilidade.
Apesar de continuar sendo uma doença genética grave, os avanços na terapêutica clínica, os novos antibióticos e os transplantes de pulmão e fígado, estão conseguindo diminuir a letalidade e aumentar a sobrevida dos pacientes, já chegando em média, aos 40 anos de idade. Diagnosticar a fibrose cística precocemente e seguir um rígido e contínuo tratamento, ainda é o melhor remédio.
