
A fibrose cística, também conhecida como mucoviscidose ou popularmente como “doença do beijo salgado”, é uma doença hereditária crônica das glândulas secretoras do muco, sucos digestivos e glândulas sudoríparas, que produzem o suor. Esses fluidos secretados são normalmente finos e escorregadios, mas em pessoas com fibrose cística, um gene defeituoso faz com que as secreções tornem-se pegajosas e espessas, aderindo em tubos, dutos e passagens.
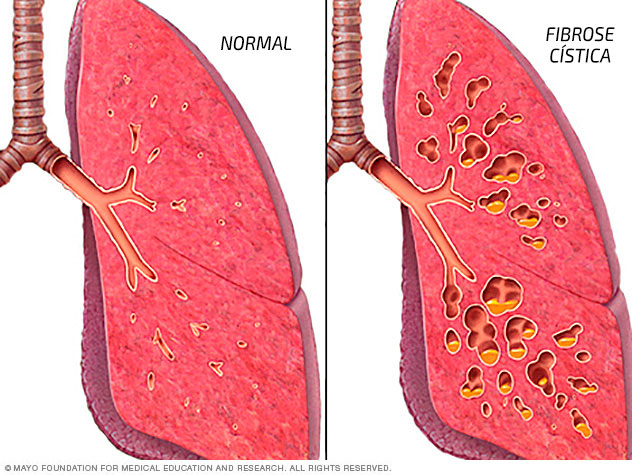
Portanto, em vez de agir como um lubrificante, as secreções se acumulam nos pulmões, pâncreas e outros órgãos como fígado, intestinos e órgãos sexuais.
Conforme dados da ABRAM - Associação Brasileira de Assistência a Mucoviscidose, a doença atinge cerca de 70 mil pessoas em todo mundo, e é a doença genética grave mais comum da infância.

Os sintomas e a severidade da fibrose cística podem variar. Algumas pessoas têm problemas sérios desde o nascimento. Outros têm uma versão mais suave da doença que não aparece até a adolescência ou vida adulta.
Nos pulmões, o muco obstrui as vias respiratórias, facilitando a multiplicação de bactérias, levando às infecções, danos pulmonares extensos e, eventualmente, insuficiência respiratória.
No pâncreas, o muco impede a liberação de enzimas digestivas, dificultando a digestão dos alimentos. Isso acontece porque as substâncias (enzimas) que digerem gorduras e proteínas não conseguem fazer a quebra enzimática dos nutrientes dos alimentos e absorver nutrientes vitais, causando desnutrição e deficiência de vitamina.
Em recém-nascidos, a doença pode causar entupimento do intestino, dificuldade de ganhar peso, tosse com secreção e desidratação sem motivo aparente.
Com o passar dos anos, o portador de fibrose cística pode apresentar perda de peso, desnutrição progressiva, sangramento pulmonar, tosse crônica com muita secreção, sinusite crônica, pneumotórax (vazamento de ar do pulmão), formação de pólipos nasais (aumento da mucosa do revestimento do nariz que formam pseudotumores), doença hepática (cirrose biliar), diabetes, infecções respiratórias, infertilidade, fezes volumosas e gordurosas, dificuldades no trânsito intestinal (evacuação, gases intestinais, barriga inchada por causa de constipação severa e dor ou desconforto abdominal, transpiração abundante, suor excessivamente salgado.

Quanto mais precoce o diagnóstico, melhor para o paciente. Atualmente o diagnóstico pode ser feito no nascimento do bebê com o teste do pezinho ou triagem neonatal, como também é conhecido o exame. Esta triagem possibilita a detecção precoce, melhorando o prognóstico e a taxa de sobrevida.
A fibrose cística também é diagnosticada através de vários exames laboratoriais, como testes de suor, sangue e genético ou de imagem.
Veja quais exames o Richet disponibiliza para o diagnóstico da fibrose cística (clique aqui)

*Imagens de tomografia computadorizada de alta resolução: 1992 (a); 2000 (b); 2015 (c)
Fonte: Monitoring clinical and microbiological evolution of a cystic fibrosis patient over 26 years: experience of a Brazilian CF Centre | Cassiana da Costa Ferreira Leite, Tania Wrobel Folescu, Mônica de Cássia Firmida, Renata Wrobel Folescu Cohen, Robson Souza Leão, Flávia Alvim Dutra de Freitas, Rodolpho Mattos Albano, Claudia Henrique da Costa and Elizabeth Andrade Marques

Não existe cura para a fibrose cística, mas os tratamentos evoluíram muito nos últimos anos e ajudam a retardar a progressão da doença.
O tipo e a gravidade dos sintomas podem diferir amplamente de pessoa para pessoa, portanto, embora planos de tratamento possam conter muitos dos mesmos elementos, eles são adaptados às circunstâncias únicas de cada indivíduo.
Os tratamentos podem incluir fisioterapia torácica, nutricional e terapias respiratórias, medicamentos e exercícios.